NIVELES DE ORDENAMIENTO ATÓMICO

NIVELES DE ENERGÍA Y ORBITALES
En un átomo los electrones ocuparán orbitales de forma que su energía sea la menor posible. Por ello se ordenan los orbitales en base a su nivel energético creciente.
La energía de los orbitales para átomos de varios electrones viene determinada por los números cuánticos n y l. En la figura de la derecha se muestran los orbitales de los 4 primeros niveles de energía (desde n = 1 hasta n = 4) y su orden de energía. Puede verse que la energía de los orbitales no coincide exactamente con el orden de los niveles. Por ejemplo, el subnivel 4s tiene una menor energía que el 3d.
Todos los orbitales de un mismo tipo que hay en un nivel tienen igual energía; por eso se colocan a la misma altura.

CELDAS UNITARIAS
Se define como celda unitaria, la porción más simple de la estructura cristalina que al repetirse mediante traslación reproduce todo el cristal. Todos los materiales cristalinos adoptan una distribución regular de átomos o iones en el espacio.
Se trata de un arreglo espacial de átomos que se repite en el espacio tridimensional definiendo la estructura del cristal. Se caracteriza por tres vectores que definen las tres direcciones independientes del sistema de coordenadas de la celda. Esto se traduce en seis parámetros de red, que son los módulos, 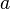 ,
,  y
y  , de los tres vectores, y los ángulos
, de los tres vectores, y los ángulos  ,
, 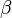 y
y 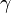 que forman entre sí. Estos tres vectores forman una base del espacio tridimensional, de tal manera que las coordenadas de cada uno de los puntos de la red se pueden obtener a partir de ellos por combinación lineal con los coeficientes enteros.
que forman entre sí. Estos tres vectores forman una base del espacio tridimensional, de tal manera que las coordenadas de cada uno de los puntos de la red se pueden obtener a partir de ellos por combinación lineal con los coeficientes enteros.
, y , de los tres vectores, y los ángulos , y que forman entre sí. Estos tres vectores forman una base del espacio tridimensional, de tal manera que las coordenadas de cada uno de los puntos de la red se pueden obtener a partir de ellos por combinación lineal con los coeficientes enteros.
La posición de un átomo dentro de la celda unidad se describe normalmente usando coordenadas fraccionarias. La simetría traslacional de una estructura cristalina se caracteriza mediante la red de Bravais, existen 14 redes de Bravais diferentes y todas las estructuras cristalinas minerales conocidas encajan en una de esas 14 disposiciones. Estas redes pueden ser:
- Tipo P: Se denomina primitiva y tiene puntos de red en los vértices de la celda.
- Tipo I: Red centrada en el interior. Esta presenta puntos de red en los vértices de la celda y en el centro de la celda.
- Tipo F: Red centrada en todas las caras. Presenta puntos de red en los centros de todas las caras, así como en los vértices.
- Tipo C: Red centrada en la base. Una red tipo C se refiere al caso en el que la simetría traslacional coloca puntos de red en los centros de las caras delimitados por las direcciones a y b así como en el origen.
No hay comentarios.:
Publicar un comentario